药学院杨悦课题组发文评估我国突破性疗法加速程序对新药研发的激励
近日,清华大学药学院杨悦研究员课题组对近年来中国获得突破性疗法资格认定(BTD)的药物进行评估,肯定了BTD程序对新药研发的激励效果。
2020年7月国家药品监督管理局组织制定《突破性治疗药物审评工作程序(试行)》,以加快用于防治严重危及生命或严重影响生存质量疾病的药物研发,引起了行业广泛关注。该研究介绍了我国突破性治疗药物程序的特点,并对自该文件发布开始到2022年4月获批的78个获得突破性疗法资格认定的新药(82个适应症)进行了评估。结果显示,获得突破性疗法资格认定的国产新药主要集中在肿瘤领域。进口新药获得突破性疗法资格认定的时间明显快于国产新药。与非BTD药物相比,BTD药物可以显著缩短临床试验和审评时间。此外,近35%的BTD药物同样获得了美国食品药品监督管理局(FDA)的突破性疗法资格认定,这表明企业可采用在中国和美国同时申请BTD的研发策略,加快新药在全球同步上市。BTD的实施有望进一步加快我国新药的研发,更好地解决未满足的临床需求。
在82个获得BTD的药物适应症中,肿瘤领域占比过半(51/82),其次为抗感染药物。在抗肿瘤BTD药物中,最常见为用于治疗淋巴瘤(14/82)和肺癌(13/82)。相比于进口BTD药物,国产BTD药物更加聚焦于肿瘤领域(34% vs. 86%; P<0.001),其中最常见的抗肿瘤靶点为PD1/PDL1(n=7),HER2(n=6)和P13K(n=6)(图1)。
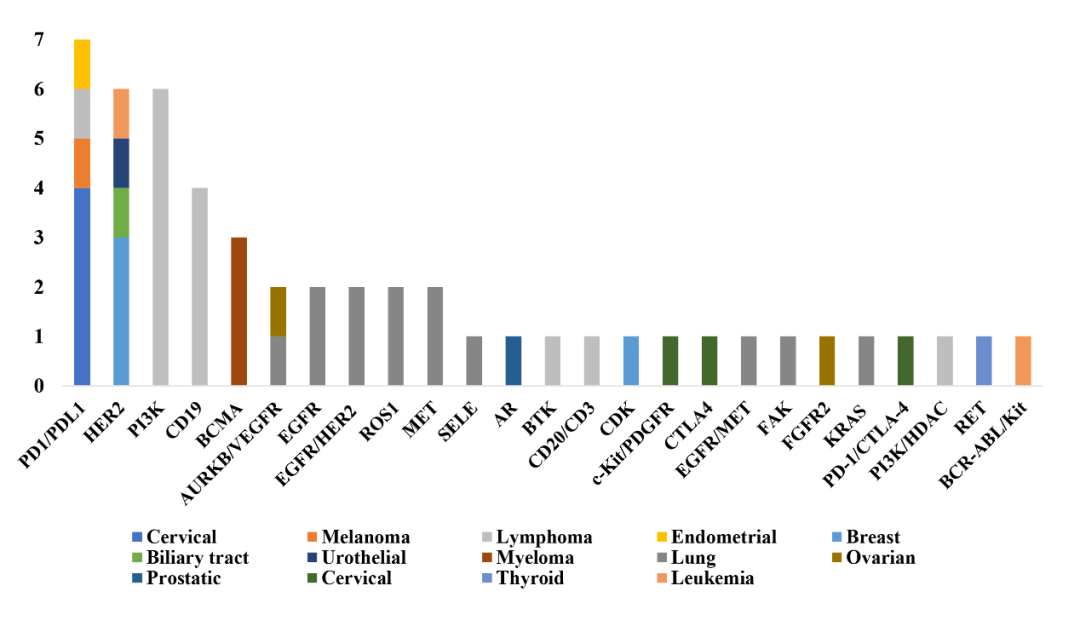
图1.抗肿瘤BTD药物靶点分布(n=51)
课题组对获得BTD药物的时间(从IND批准时间至BTD批准时间)进行了单因素影响分析,发现获得BTD时间与是否为国产或进口新药相关,而与给药途径(口服或非口服)、药物类型(小分子药物或生物制品)、药物分类(新药或改良新药)以及是否获得FDA的BTD无明显关系(图2)。这可能与我国2018年开始接受境外临床试验数据有关。由于部分进口新药在进入我国开展临床试验之前已在境外积累了一些临床试验数据,因此相比国产新药可能更快获得我国的突破性疗法资格认定。
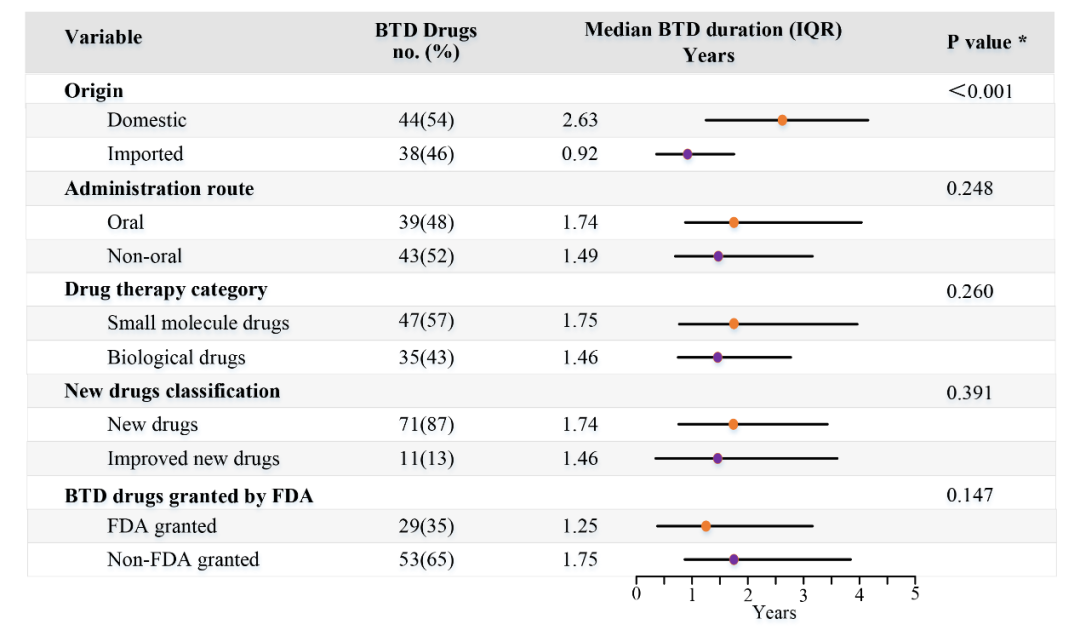
图2.获得BTD药物时间的单因素影响分析
课题组以2021年批准的1类新药(非BTD药物)作为对照组,对比了BTD药物和非BTD药物在临床试验时间(定义:IND批准时间到NDA提交时间)以及审评时间方面(定义:NDA提交时间到NDA批准时间)的差异。结果显示BTD药物可显著缩短临床试验及审评时间(图3)。由于纳入的BTD药物数量有限,该结论有待进一步验证。
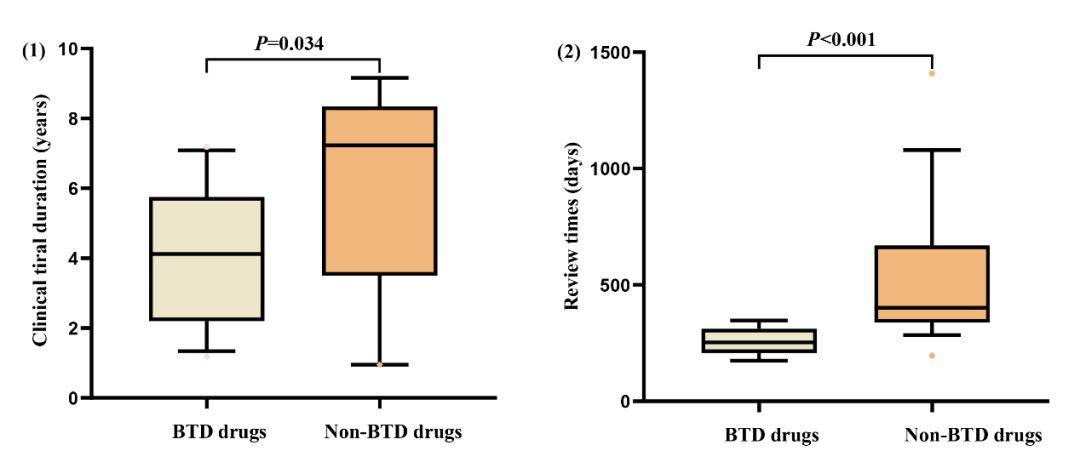
图3.对比BTD药物和非BTD药物在临床试验时间和审评时间方面的差异
近年来,有不少国外研究发现BTD药物在有效性、安全性和创新性方面与非BTD药物并无明显差异,这也提示我国应当逐步完善BTD标准,包括加强对生物标志物和替代终点研究、建立临床价值评估框架等,以保证BTD药物有真正的突破性治疗效果,为患者带来更多的希望。
本研究论文题目为“中国获得突破性疗法资格认定的药物评估:2020-2022年汇总分析”(Assessment of the breakthrough-therapy-designated drugs granted in China: a pooled analysis 2020–2022),应邀在线发表于药学知名期刊《今日药物发现》(Drug Discovery Today)。论文第一作者为清华大学药学院博士后罗兴献,通讯作者为杨悦,药学院钱锋院长课题组对该研究提供了帮助和支持。该研究得到了清华-北京大学生命科学联合中心基金的项目经费支持。
【免责申明】本专题图片均来源于学校官网或互联网,若有侵权请联系400-0815-589删除。