华西医院生物国重陈崇团队揭示小细胞肺癌转移的表观遗传学重编程分子调控机制
小细胞肺癌(SCLC)是一类恶性程度极高的神经内分泌肿瘤,发病率约占肺癌总数的15-20%,其5年生存率不到10%,极强的远端转移能力是SCLC的显著特征。我校华西医院生物治疗国家重点实验室陈崇教授团队在Nature Cancer发表研究论文”KMT2C deficiency promotes small cell lung cancer metastasis through DNMT3A-mediated epigenetic reprogramming”,阐明了组蛋白甲基转移酶KMT2C缺失通过组蛋白-DNA协同低甲基化促进SCLC远端转移的表观遗传学机制,对SCLC治疗具有一定的理论意义。

远端转移是癌症致死的主要原因。而SCLC是人类肿瘤中转移能力最强的肿瘤之一,在诊断时,超过70%的SCLC患者都已经发生了转移,因而不能进行手术治疗,进而也导致研究材料的缺乏。为此,陈崇教授团队创建了一类新型的“精准肿瘤模型”。团队利用最新的肺类器官培养、基因编辑和原位移植等技术,构建了原发、原位、驱动基因明确的SCLC小鼠模型。该模型与临床患者的病理特征、基因表达等高度相似,同时表现出广泛的侵袭转移能力。
基于该SCLC模型,研究团队通过单细胞测序以及临床大数据协同分析,构建了SCLC转移的分子路径,发现KMT2C是一个关键的SCLC转移基因。KMT2C,又叫MLL3,是组蛋白H3的4位赖氨酸1-、2-甲基转移酶,在多种人类肿瘤中高频突变。前期陈崇教授首次证明KMT2C在白血病中是一个单倍剂量不足的肿瘤抑制基因(2)。本研究中,团队发现KMT2C的表达沿着SCLC的转移路径逐渐下调,并在有远端转移的SCLC患者中有更高频率的突变。研究团队利用体外类器官和小鼠体内功能学实验证明KMT2C的敲除显著地促进SCLC的进展和远端转移。
进而,团队利用表观遗传学相关的多组学联合分析和生物学功能研究,阐明了KMT2C通过直接调控DNMT3A表达对SCLC细胞表观遗传学重编程的分子机制。KMT2C缺失引起是组蛋白H3的4位赖氨酸1-、2-甲基化水平的显著下调,从而影响下游基因的表达。DNA甲基转移酶DNMT3A是KMT2C直接调控的下游基因之一。因而,KMT2C缺失的SCLC也呈现出显著的DNA低甲基化。而回补DNMT3A可以抑制KMT2C缺失SCLC的转移。特别有意思的是,DNA和组蛋白甲基转移酶的共同底物S-腺苷甲硫氨酸可以提高KMT2C缺失的SCLC中的DNA和组蛋白甲基化水平,从而显著的抑制肿瘤的进展与转移。
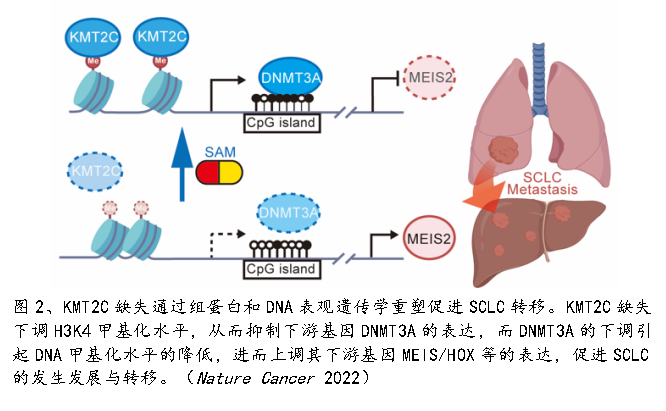
该工作的科学意义和创新性在于一是研究团队利用肺类器官培养、基因编辑和原位移植等技术,构建了原发、原位、驱动基因明确的SCLC小鼠模型。二是研究团队通过单细胞多组学技术、多种表观遗传学协同分析平台、临床大数据分析以及体内/体外的功能学实验,发现KMT2C是一个SCLC转移相关的关键基因。三是阐明了KMT2C缺失直接下调了DNMT3A造成了组蛋白-DNA协同低甲基化,对SCLC细胞的表观遗传学进行了重编程,从而促进SCLC远端转移发生的分子机制。四是阐明SAM可以作为KMT2C突变SCLC患者的新型治疗方案,用于抑制肿瘤转移的发生和恶性进展。研究团队期待KMT2C可以作为一个新的分子标记物为SCLC患者的临床诊断提供重要参考。也期望SAM可以作为一种新的治疗方式提高KMT2C突变患者的生存时间和生存质量。此外,团队自主构建的新型SCLC小鼠模型也将广泛地应用于SCLC的基础与转化研究。
四川大学华西医院肿瘤中心胸部肿瘤科纳飞飞博士、陈婧瑶博士、生物治疗国家重点实验室博士研究生潘翔宇、陈雪兰为文章的共同第一作者,陈崇教授为通讯作者。该工作得到了魏于全院士的指导。卢铀教授、刘玉教授等参与了研究。国家自然科学基金、科技部重点专项、四川大学华西医院等提供了经费支持。
陈崇教授团队长期研究肿瘤表观遗传学机制。通过构建血液肿瘤和多种实体肿瘤原发原位的动物模型,探究肿瘤发生发展的分子机制,进而基于机制和高通量筛选鉴定治疗靶标和药物。前期发现了KMT2C、PHF23等多个表观遗传学肿瘤抑制基因,相关工作发表在Nature、Cancer Discovery、Cancer Cell、STTT等期刊。
【免责申明】本专题图片均来源于学校官网或互联网,若有侵权请联系400-0815-589删除。