西安交大科研人员开发出高效的宏基因组分析新工具
细菌、古细菌等微生物作为地球上现存最古老的生命形式,在生物进化、生命健康、物质循环和环境变化的进程中扮演重要角色。绘制完整基因组序列是研究其系统分类的基础,但受限于微生物基因组序列的复杂性,基于测序技术和序列拼装算法从混合微生物样本中获得完整基因组序列非常困难,尤其对于不可培养的稀有细菌,迄今仅有少量的微生物被测序。因此,急需在聚类方法上突破,开发出从混合微生物序列集(宏基因组)中精准分类的有效工具。
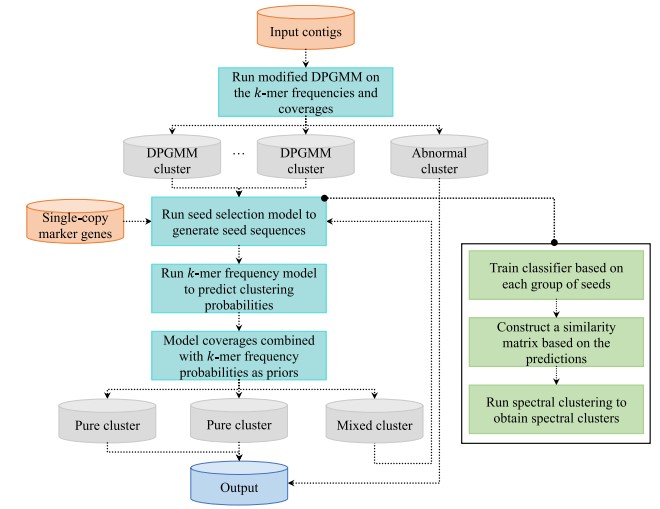
近日,西安交通大学生命学院生物医学信息与基因组学中心(Biomedical Informatics & Genomics Center, BIGC)杨铁林教授团队在宏基因组序列聚类方法上取得重要进展,开发出新的工具MetaDecoder,该工具首先构建了基于GPU的样本加权狄利克雷过程高斯混合模型(DPGMM)以降低原始宏基因组复杂性,然后使用微生物序列碱基组成模型和测序数据覆盖度模型迭代处理低复杂度宏基因组序列,最终产生完整性高且污染度低的基因组水平序列集合,此方法在准确性及计算效率上领先于当前主流的宏基因组序列聚类方法。
该项研究成果以《MdtaDecoder:一个宏基因组序列聚类新方法》(MetaDecoder: a novel method for clustering metagenomic contigs)为题在国际著名学术期刊《微生物组》(Microbiome,)上发表。该工作由西安交通大学独立完成,博士生刘聪聪为该论文第一作者,杨铁林教授为通讯作者。生物医学信息工程教育部重点实验室为第一单位,西安交通大学生命学院和第二附属医院为该论文的通讯单位。
杨铁林教授团队长期致力于生物信息学和疾病遗传机制研究,一系列成果发表在《自然综述-内分泌》(Nature Reviews Endocrinology)、《基因组医学》(Genome Medicine)、《美国人类遗传学》(American Journal of Human Genetics)等期刊。该项研究工作得到了国家自然科学基金、陕西省自然科学金、中央高校基本科研业务费专项资金等项目的资助。
【免责申明】本专题图片均来源于学校官网或互联网,若有侵权请联系400-0815-589删除。